I Craig Henderson, MD |
EDITED COMMENTS |
 The role of adjuvant aromatase inhibitors in postmenopausal women The role of adjuvant aromatase inhibitors in postmenopausal women
Based on data from various adjuvant endocrine therapy trials, I believe it is unreasonable to withhold aromatase inhibitors from postmenopausal women with hormone receptor-positive disease. ATAC is still the definitive adjuvant trial in terms of comparing tamoxifen to an aromatase inhibitor, and the data are very compelling (ATAC Trialists’ Group 2005; Howell 2004). An aromatase inhibitor is now my drug of choice and that changed in just the past years.
Having said that, I’m not certain that the last “shoe has dropped.” We have not yet seen a survival benefit with aromatase inhibitors, and it’s possible that the late effects may be different than the early effects. I’m not prepared to completely abandon the SERMs in adjuvant therapy.
It is also quite plausible that optimal endocrine therapy will vary from patient to patient, and there may be a subset that benefits more from tamoxifen while another benefits more from withdrawal of estrogen, which is what we accomplish with aromatase inhibitors.
In addition, we now have selective estrogen receptor downregulators (SERDs) like fulvestrant on the horizon. While we don’t have data yet in the adjuvant setting, I imagine it won’t be long before we begin to see data with these agents. As for switching patients from tamoxifen to an aromatase inhibitor, I discuss this with every postmenopausal patient on tamoxifen. My tendency, which is based on my intuition rather than data, is to advise patients on tamoxifen to complete two or three years and then switch. We don’t know the optimal time to switch, and we don’t know the optimal duration of various endocrine therapies. While we know that five years of tamoxifen is as good or better than 10 years, the optimal duration of aromatase inhibitors is unknown at this time.
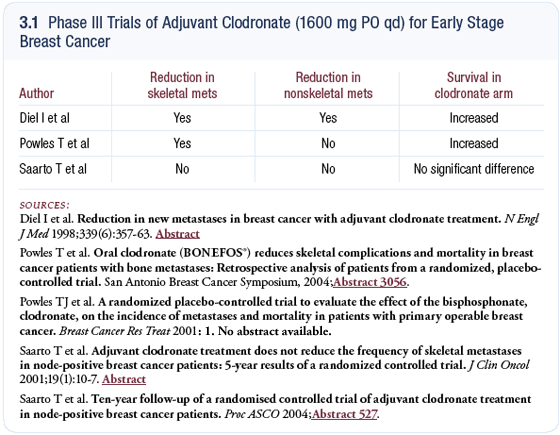
Anticancer effect of bisphosphonates
It is possible that when we introduce a bisphosphonate to reduce bone loss, we may be introducing an anticancer treatment as well. I find Ingo Diel’s hypothesis very compelling. Enough circumstantial evidence exists that I believe we should seriously consider the possibility that bone mediates metastases in breast cancer and, therefore, the state of the bone will indirectly impact survival. There’s no question that much of the data suggests a decrease in breast cancer mortality when one uses a bisphosphonate (Diel 1998, 2000); [3.1].
Surrogate endpoints to detect effects of new agents
We’re moving into an era in which we’re beginning to combine biologic agents with chemotherapy or other biologic agents. Investigators from UCLA presented data at the 2004 San Antonio Breast Cancer Symposium examining trastuzumab plus bevacizumab and showed responses with the combination that couldn’t be achieved with trastuzumab alone (Pegram 2004). It’s a small study, but it gives us a clue as to where we are headed in breast cancer treatment.
I’ve been trying to figure out how we can learn about drug interactions when combining agents without conducting large randomized trials. There are biological agents that have a synergistic effect when combined with conventional cytoxics or have some effect by themselves.
There are also agents with only synergistic effects and none on their own. In randomized trials with real patients, such agents may be quickly abandoned if no positive effects are seen.
Somehow we’ve got to keep clinicians and patients excited about new drugs and find something that signals us to take them to the next level. I believe our best hope right now is to use surrogate endpoints such as PET scanning, which is being used by some investigators. With PET scans we can measure the effects of therapy on proliferation very early and may be able to inhibit proliferation without actually inducing apoptosis.
Amplifying weak signals detected in clinical trials
The initial Phase I trastuzumab trial was conducted by Dennis Slamon at UCLA. Then UCSF and Memorial Sloan-Kettering jointly conducted the Phase II study. When we submitted the Phase II data to the New England Journal of Medicine, with a response rate just over 11 percent, they said it wasn’t positive enough and rejected it. These were studies conducted by well-known investigators at top institutions, but the Journal felt it was premature.
Personally, the trastuzumab study was probably the most important and exciting thing I’ve done in my career. We had a patient at UCSF with massive ascites and large liver metastases who had progressed on three different types of chemotherapy. When she received single-agent trastuzumab, she had a 50 percent response rate and no toxicities. Her ascites disappeared and her quality of life improved. We had to look beyond the 11 percent response rate, because we could see that for some patients it truly worked.
When we see such responses despite a weak signal, we need to determine how to make that signal larger. If we had not targeted the population with HER2-positive disease, it would have taken almost 30 years to complete the trastuzumab study with approximately 10,000 women, and the positive effect would have been diluted because most tumors do not overexpress HER2.
By examining the population with HER2-positive disease alone, we amplified the signal and found that trastuzumab reduces mortality by 25 percent in the worst form of the disease. The challenge now is to take these weak signals — drugs with only five percent response rates — and determine how to amplify them.
Early in my career, I was warned that whereas a false positive will get sorted out, a false negative could bury a drug forever. In cancer we can’t afford to do that. Trastuzumab was a near miss and if we hadn’t seen the positive effects, I don’t think we’d be examining other targeted agents like gefitinib, bevacizumab or erlotinib. Gefitinib is another example where we saw only a 10 percent response rate in randomized trials, but fortunately physicians who were paying attention to clinical results kept it alive and kept the whole field of targeting EGFR alive as well.
AKT inhibitors and targeted therapy
Currently I’m excited about the development of an AKT inhibitor, perifosine (KRX-0401). Researchers at a small German company first identified this agent’s antiproliferative and apoptotic effects. Sausville and his colleagues at the NCI showed that this agent inhibits AKT and since then three or four other labs have demonstrated this as well (Patel 2002). Currently it’s the only drug that’s in the clinic that is known to have this pronounced effect on AKT.
While we do not yet have proof of principle that the inhibition of AKT is the mechanism of action for perifosine, we know it affects MAP kinase and p21waf1/cip1 and has multiple effects in the cell. At the beginning of my career, I really believed that the way we would cure cancer was related to dose, but I no longer believe that. I’m keen on drugs that target new areas of the cancer cell that we couldn’t target five, 10 or 20 years ago. This drug looks like it has that potential, and it has an antitumor effect that’s well documented.
Numerous challenges exist when developing new agents, such as determining which patients to target and how to characterize them and determining the ideal doses to be used.
Select Publications
 |
Dr Henderson is an Adjunct Professor of Medicine at the University of California in San Francisco, California. |
|
